Klinefelter sendromu, bir erkeğin fazladan bir X kromozomu ile doğduğu genetik bir durumdur. Klinefelter sendromlu erkekler tipik XY kromozomları yerine XXY’ye sahiptirler, bu nedenle bu duruma bazen XXY sendromu denir.
Nedenleri
Klinefelter sendromu, bir erkeğin fazladan bir cinsiyet kromozomu ile doğmasına neden olan rastgele bir hatanın sonucu olarak ortaya çıkar. Kalıtsal bir durum değildir.
Bu kromozom, testislerin gelişimine müdahale eden ve normalden daha az testosteron (erkek cinsiyet hormonu) ürettikleri anlamına gelen fazladan gen kopyaları taşır.
İnsanlar tipik olarak her hücrede ikisi cinsiyet kromozomu olan 46 kromozoma sahiptir. Dişilerde iki X kromozomu (46, XX) ve erkeklerde bir X ve bir Y kromozomu (46, XY) bulunur. Çoğu zaman, Klinefelter sendromlu erkekler, toplam 47 kromozom için olağan X ve Y kromozomlarına ek olarak fazladan bir X kromozomuna sahiptir (47, XXY).
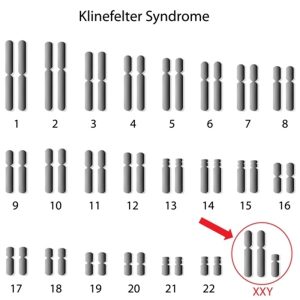
Klinefelter sendromu özelliklerine sahip bazı kişiler, hücrelerinin sadece bazılarında fazladan bir X kromozomuna sahiptir; diğer hücreler tipik olarak bir X ve bir Y kromozomuna sahiptir. (Nadiren, diğer hücrelerde ek kromozom anormallikleri olabilir.) Bu bireylerde durum mozaik Klinefelter sendromu (46, XY / 47, XXY) olarak tanımlanır. Klinefelter sendromlu bireylerin yüzde 10’undan daha azının mozaik forma sahip olduğu düşünülmektedir. Mozaik Klinefelter sendromlu erkeklerin, tüm hücrelerinde fazladan X kromozomu olanlardan daha hafif belirti ve semptomları olabilir.
Her hücrede birden fazla ekstra cinsiyet kromozomunun varlığından kaynaklanan çeşitli koşullar, bazen Klinefelter sendromunun varyantları olarak tanımlanır. Bu koşullar 48, XXXY sendromu , 48, XXYY sendromu ve 49, XXXXY sendromunu içerir . Klinefelter sendromu gibi, bu koşullar erkek cinsel gelişimini etkiler ve öğrenme güçlükleri, konuşma ve dil gelişimi ile ilgili problemlerle ilişkilendirilebilir. Bununla birlikte, bu bozuklukların özellikleri Klinefelter sendromundan daha şiddetli olma eğilimindedir ve vücudun daha fazla bölümünü etkiler.
Semptomlar
Klinefelter sendromu genellikle erken çocukluk döneminde belirgin semptomlara neden olmaz ve daha sonraki semptomların fark edilmesi zor olabilir.
Her zaman mevcut olmayan olası özellikler şunları içerebilir:
- bebeklerde ve yeni yürümeye başlayan çocuklarda – normalden daha geç oturmayı, emeklemeyi, yürümeyi ve konuşmayı öğrenmek, normalden daha sessiz ve daha pasif olmak
- çocuklukta – utangaçlık ve düşük özgüven, okuma, yazma, heceleme ve dikkat etme sorunları, hafif disleksi veya dispraksi , düşük enerji seviyeleri ve sosyalleşmede veya duyguları ifade etmede zorluk
- gençlerde – aile için beklenenden daha uzun (uzun kollar ve bacaklar), geniş kalçalar, zayıf kas tonusu ve normalden daha yavaş kas büyümesi, normalden daha geç büyümeye başlayan yüz ve vücut kıllarında azalma, küçük bir penis ve testisler, büyümüş göğüsler (jinekomasti)
- yetişkinlikte – yukarıda belirtilen fiziksel özelliklere ek olarak doğal olarak çocuk sahibi olamama (kısırlık) ve düşük cinsel dürtü
Teşhis
47, XXY (KS) olan erkekler genellikle doğumdan önce (örneğin kromozomal bozukluklar için doğum öncesi taramalar yoluyla), ergenlik döneminde veya düşük doğurganlık nedeniyle yaşamın ilerleyen dönemlerinde tanımlanır. 47, XXY (KS), bir kan numunesi üzerinde bir kromozom karyotip analizi veya bir kromozom mikrodizi (CMA) testi ile teşhis edilir. CMA, oral yanak (bukkal) sürüntüden oluşur ve kromozom sayılarındaki anormallikleri saptamanın ve kesin tanı sağlamanın kolay ve ağrısız bir yoludur. 47, XXY (KS) ayrıca koryonik villöz veya amniyotik sıvı hücrelerinde amiyosentez ile prenatal olarak teşhis edilebilir.
Tedavi
Klinefelter sendromunun ayırt edici özelliklerinden biri, testosteron eksikliğine neden olan bir durum olan hipergonadotropik hipogonadizmdir. Tedavi, testosteron enantat, cypionate veya androgel gibi erkek hormonlarının (androjenler) hedefli uygulanmasını içerir. Erken hormon tedavisi (EHT), üç ayda bir 25 mg testosteron enantat enjeksiyonu, tipik olarak 4-12 aylıkken uygulanır. Bu hormonlar, ikincil erkek cinsel özelliklerinin gelişimini (virilizasyon) teşvik etmek ve yetersiz testosteron seviyeleri nedeniyle meydana gelen feminizasyon etkilerini hafifletmek için verilir. Hormon replasman tedavisi erken bebeklik döneminde veya pubertal gelişim sırasında veya hatta yaşamın ilerleyen dönemlerinde başlatıldığında etkilidir.
Bebekken, bu çocukların pediatrik fizik tedavi ile tedavi edilebilen pozisyonel tortikolis açısından izlenmesi gerekir. Konuşma ve dil terapisi, fizik tedavi ve mesleki terapi genellikle 47, XXY (KS) olan erkekler için yararlıdır. Bu müdahalelerin 47, XXY (KS) olan erkek çocuklarda akademik, fiziksel, bilişsel ve sosyal sonuçları önemli ölçüde iyileştirdiği gösterilmiştir. Sosyal beceri eğitimi dersleri de faydalı olabilir.
47, XXY (KS) olan erkekler düşük doğurganlığa sahiptir ve yeni yardımcı üreme teknikleriyle, daha fazla erkek çocuk üreme fırsatına sahiptir. Mozaik 47, XXY (KS) olan erkeklerin üreme ile daha az komplikasyon olasılığı daha yüksektir. Testislerden spermin cerrahi olarak çıkarılması ve intrasitoplazmik sperm enjeksiyonu (ICSI), 47, XXY (KS) olan erkeklere yardımcı olmak için mevcut tıbbi bir teknolojidir.


Yorum ekle